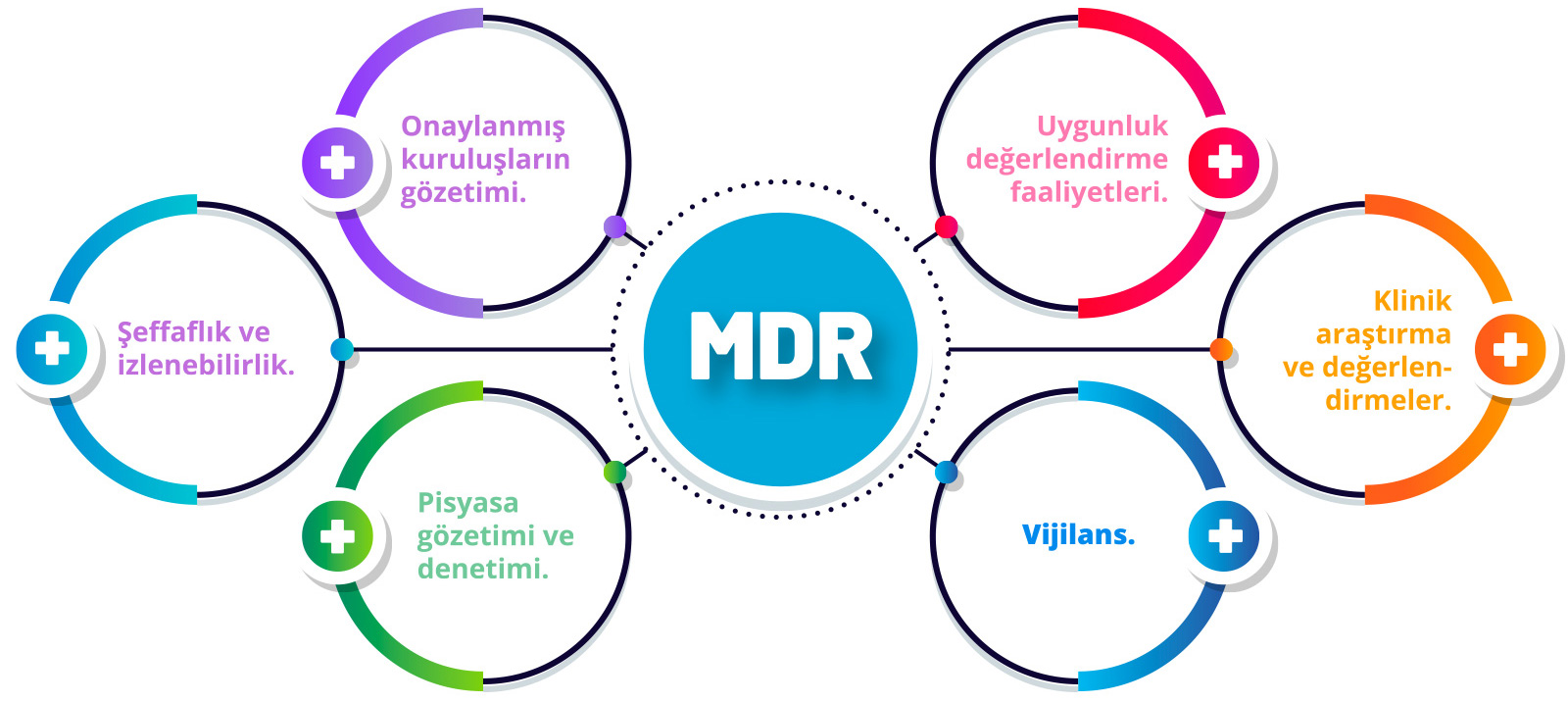
Bir tıbbi cihazın CE işareti (Conformité Européenne veya Avrupa Uygunluğu) için uygunluğunun değerlendirilmesi,
her bir tıbbi cihaz için risk sınıfına göre değişir. Risk sınıflandırmasının yanı sıra, örneğin bir tıbbi cihazın
steril olması gerektiğinde, belirli özellikler uygunluk değerlendirme prosedürünü etkileyebilir.
Tüm Sınıf IIa, IIb ve III tıbbi cihazların yanı sıra bazı özel Sınıf I cihazlar, bir Onaylanmış Kuruluşun müdahalesini gerektirir
(MDR Madde 52 (7)(a6, b7, c8). MDR Ekleri IX, X ve XI, cihazın sınıfına göre farklı değerlendirme yollarını tanımlar.
Bazı durumlarda üreticiler, Yönetmelikte açıklanan birkaç seçenek arasından uygunluk değerlendirme yolunu seçebilir.
Sınıf III implante edilebilir cihazlar ve belirli Sınıf IIb cihazlar için bağımsız bir uzman paneli tarafından
yürütülecek yeni bir klinik değerlendirme danışma prosedürü vardır. Onaylanmış Kuruluş, uzman paneli tarafından
ifade edilen bilimsel görüşü dikkate almak zorunda olacaktır (MDR Madde 54).
2001/83/AT sayılı Direktif’ i, (AT) 178/2002 sayılı Tüzük’ ü ve (AT) 1223/2009 sayılı Tüzük’ ü değiştiren ve
90/385/AET ve 93/42/AET sayılı Konsey Direktiflerini yürürlükten kaldıran, tıbbi cihazlara ilişkin 5 Nisan
2017 tarihli ve (AB) 2017/745 sayılı AVRUPA PARLAMENTOSU VE KONSEY TÜZÜĞÜ'dür.
93/42/AET (MDD) ve 90/385/AET (AIMDD) Direktiflerinden (AB) 2017/745 sayılı Tüzük (MDR)’e Geçiş Sebepleri;
Bu Tüzük, hastalar ve kullanıcılar için sağlığın yüksek seviyede korunmasını esas alarak ve bu sektörde faaliyet
gösteren küçük ve orta ölçekli işletmeleri dikkate alarak, tıbbi cihazlarla ilgili iç piyasanın aksamadan işlemesini
sağlamayı amaçlar. Aynı zamanda bu Tüzük, bu tür ürünlerle ilgili ortak güvenlik kaygılarını karşılamak üzere tıbbi
cihazlar için yüksek kalite ve güvenlilik standartları belirler. Her iki hedef eş zamanlı gerçekleştirilir ve
biri diğerine ikincil olmayacak şekilde birbiriyle ayrılmaz şekilde bağlantılıdır.
MDR tam metnine ulaşmak için tıklayınız.
MDR Eklerine ulaşmak için tıklayınız.
Genel olarak, MDR, Direktifin tüm gerekliliklerini korurken, kendi bazı yeni gerekliliklerini de ekler. Eski yönetmelik ile
karşılaştırıldığında, yeni Yönetmelik, klinik verilerle desteklenen bir yaşam döngüsü yaklaşımını vurgulamaktadır. Yönetmelik,
Onaylanmış Kuruluşların atanması için daha katı kurallar ekler. Ulusal yetkili makamlar ve Komisyon için daha fazla kontrol
ve izleme gerekliliği getirir. Yeni Yönetmelik, imalatçıların, yetkili temsilcilerin, ithalatçıların ve dağıtıcıların
yükümlülüklerine açıklık getirmektedir. MDR, belirli cihazları yeniden sınıflandırır ve eski yönetmelikten daha geniş
bir kapsama sahiptir. Bazı yüksek riskli tıbbi cihazlar için ek bir pazar öncesi konsültasyon prosedürü getirmektedir.
Diğer Direktiflerle düzenlenen ürünler (gıda, kozmetik, tıbbi ürünler, kan ürünleri vb.) ve İleri tedavi ürünleri
ve canlı materyaller içeren ürünler kapsam dışı bırakılmıştır.
Ek XVI Tıbbi amaçlı olmayan ürünler eklenmiştir.
Renkli kontact lensler.
Liposakşın.
Dermal dolgular.
IPL, epilasyon lazerleri.
Dövme silme ekipmanları.
'tıbbi cihaz' tanımına:
Hastalığın tahmini, prognozu,
Fizyolojik ya da patolojik sürecin veya durumun araştırılması, ikame edilmesi veya modifikasyonu,
İnsan vücudundan elde edilen örneklerin in vitro tetkiki vasıtasıyla bilgi sağlanması amaçları,
İmplantlar ile reaktifler,
Gebeliğin desteklenmesine yönelik cihazlar,
Tıbbi cihazların, cihaz aksesuarlarının ve Ek XVI’daki ürünlerin temizliği, dezenfeksiyonu veya sterilizasyonu için özel olarak tasarlanan ürünler
eklenmiştir.
‘aksesuar’ tanımına:
Tıbbi cihazın/cihazların kullanım amacı/amaçları bakımından tıbbi işlevselliğine doğrudan ve
spesifik olarak yardımcı olmak için kullanılan parçalar
eklenmiştir.
Tıbbi Cihaz Yönetmeliği (MDR) kapsamına giren cihazlar, imalatçı tarafından tek başına veya kombinasyon halinde
aşağıdaki özel tıbbi amaçlarla insanlar için kullanılması amaçlanan herhangi bir araç, aparat, alet,
yazılım, implant, reaktif, malzeme veya diğer eşyalardır:
Hastalığın teşhisi, önlenmesi, izlenmesi, öngörüsü, tahmini, tedavisi veya hafifletilmesi,
Bir yaralanma veya sakatlığın teşhisi, izlenmesi, tedavisi, hafifletilmesi veya tazmin edilmesi,
Anatominin veya fizyolojik veya patolojik bir sürecin veya durumun araştırılması, değiştirilmesi veya değiştirilmesi,
Organ, kan ve doku bağışları da dahil olmak üzere insan vücudundan elde edilen örneklerin in
vitro incelenmesi yoluyla bilgi sağlanması. Ve asıl amaçlanan etkisini farmakolojik, immünolojik veya metabolik
yollarla gerçekleştirmeyen, ancak işlevine bu tür yollarla yardımcı olunabilen.
Aşağıdaki ürünler de tıbbi cihaz olarak kabul edilir:
Gebeliğin kontrolü veya desteklenmesi için cihazlar,
Cihazların temizlenmesi, dezenfeksiyonu veya sterilizasyonu için özel olarak tasarlanmış ürünler,
Bazı estetik cihazlar artık Tıbbi Cihaz Yönetmeliği (MDR) kapsamına girmektedir,
Tıbbi cihaz aksesuarları tıbbi cihaz olarak kabul edilir,
Hem tıbbi hem de tıbbi olmayan kullanım amacına sahip cihazlar, gereklilikleri kümülatif olarak karşılamalıdır.
Ayrıca, Ek XVI, aşağıdaki ürün gruplarının MDR gerekliliklerine uymasını zorunlu kılar:
Göz içinde veya üzerinde kullanılan kontakt lensler ve diğer ürünler (göz damlaları ve kozmetik kontakt lensler buraya dâhil edilecektir).
Anatomiyi değiştirmek için cerrahi olarak invaziv yollarla vücuda sokulan ürünler (silikon meme implantları artık burada kabul edilecektir).
Yüz veya diğer deri altı dolgular için kullanılan ürün ve maddeler.
Liposuction (yağ aldırma), lipoliz veya lipoplasti için kullanılan ekipman.
Dövme ve epilasyon için kullanılan yüksek yoğunluklu radyasyon ekipmanı.
Beyni uyarmak için elektrik veya manyetik akım kullanan ekipman.
Aksesuarlar:
Aksesuarlar, bir cihazın kullanım amacına uygun olarak kullanılmasını sağlayan veya tıbbi cihazın tıbbi işlevselliğine yardımcı olan ürünlerdir.
MDD gerekliliklere benzer şekilde MDR kapsamında da yer almaktadırlar. Bazı ürünler artık tıbbi cihaz tanımına eklendiğinden aksesuarları da
Yönetmelik kapsamına girecek. Bu nedenle, aksesuarları MDR'de tanımlandığı şekilde tanımlamak için bu yeni tıbbi cihaz kategorileriyle birlikte
kullanılan tüm ürünlerin doğrulanması gerekir.
Bazı örnekler:
Sterilizasyon ağları.
İnvaziv veya implante edilebilir kontraseptifleri konumlandırmak veya çıkarmak için kullanılan cihazlar.
Göğüs implantlarının beklenen etkilerinin görüntülerini oluşturmak için kullanılan yazılım.
Borderline (sınırda, belirsiz) Cihazlar
Tıbbi cihazlar alanında Avrupa Komisyonu'nun (EC) bir danışma organı olan Tıbbi Cihaz Koordinasyon Grubu'nun (MDGC) bir alt grubu
olan Borderline ve Sınıflandırma Çalışma Grubu (BCWG), borderline ve sınıflandırma konusunda bir kılavuz yayınladı.
MDCG tarafından yayınlanan kılavuz belgeler aşağıdaki gibidir:
Manual on borderline and classification under Regulations (EU) 2017/745 and 2017/746 v2
MDCG 2022-05 Guidance on borderline between medical devices and medicinal productsunder Regulation (EU) 2017/745 on medical devices
AB MDR ve IVDR'ye göre ortak spesifikasyonların tanımı, "bir cihaz, süreç veya sistem için geçerli yasal yükümlülüklere
uymanın bir yolunu sağlayan, bir standart dışında teknik ve/veya klinik gereksinimler seti" şeklindedir.
Ortak özellikler kavramı, AB MDR'nin 9. Maddesinde açıklanmaktadır:
[….], uyumlaştırılmış standartların olmadığı veya ilgili uyumlaştırılmış standartların yetersiz olduğu durumda veya kamu
sağlığı sorunlarının ele alınmasına ihtiyaç duyulduğu durumda, Komisyon, MDCG’ye danıştıktan sonra, uygulama tasarrufları
vasıtasıyla, I. Ekte belirtilen genel güvenlilik ve performans gereklilikleri, II. ve III. Eklerde belirtilen teknik
dokümantasyon, XIV Ekte belirtilen klinik değerlendirme ve piyasaya arz sonrası klinik takip veya XV. Ekte belirtilen
klinik araştırmaya ilişkin gereklilikler hakkında ortak spesifikasyonlar kabul edebilir.
Komisyon tarafından oluşturulan ilk Ortak Spesifikasyonlar, tek kullanımlık tıbbi cihazların yeniden işlenmesi için oluşturuldu.
Daha fazla bilgi edinmek için tıklayınız
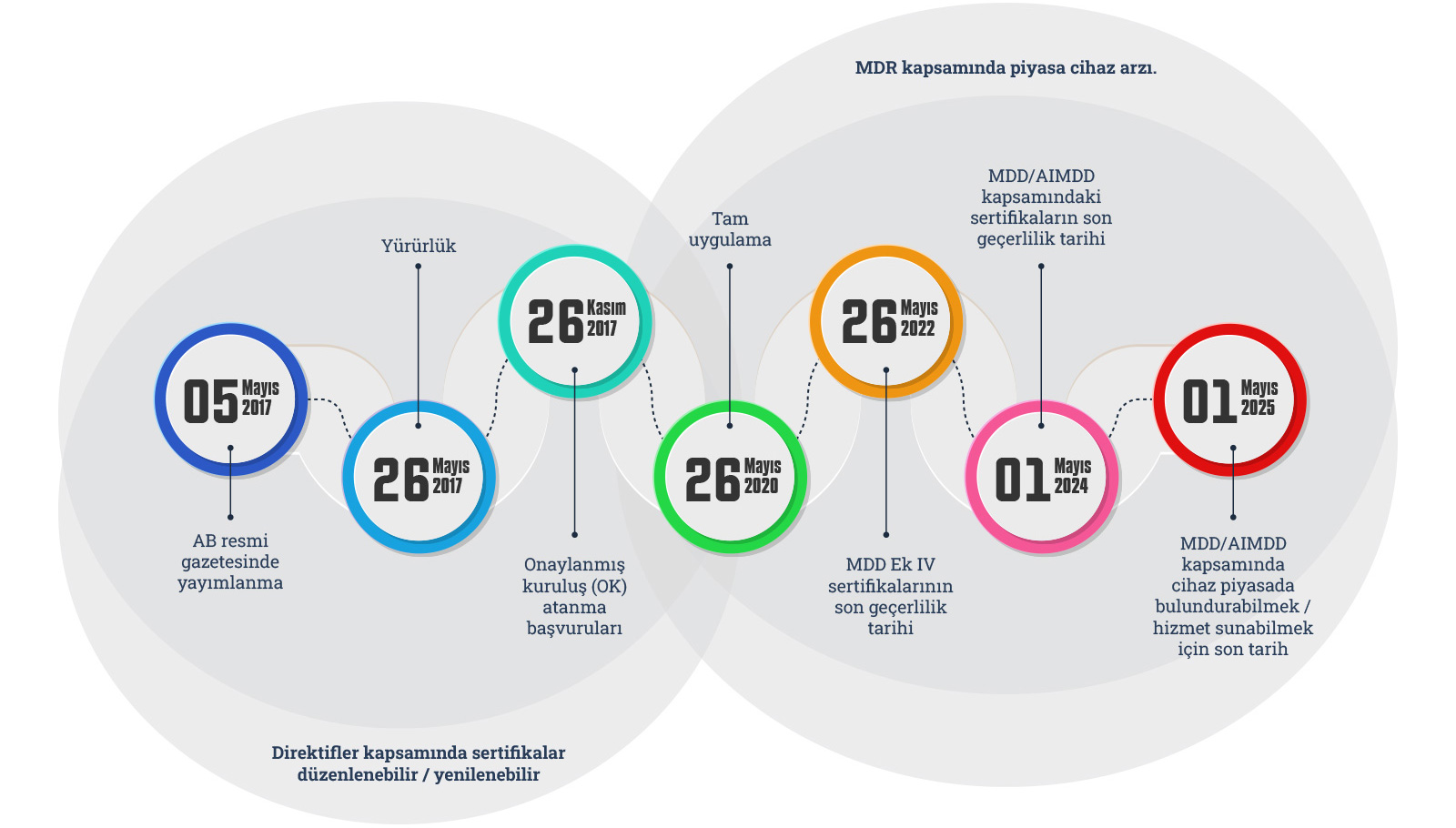
Normal şartlarda yukarıdaki gibi işleyen geçiş süreç ile ilgili olarak Komisyon 6 Ocak 2023 tarihinde, kısıtlı süre riskini azaltmak sebebiyle tıbbi cihazların onaylanması için daha fazla zaman verilmesi teklifini kabul etti. Teklif, Tıbbi Cihazlar Yönetmeliği'nde öngörüldüğü gibi, yeni kurallara uyum sağlamak için daha uzun bir geçiş dönemi getiriyor. Yeni son tarihler, tıbbi cihazların risk sınıfına bağlıdır ve hastaların tıbbi cihazlara sürekli erişimini sağlayacaktır. Ayrıca, mevcut yasal çerçeveye uygun olarak piyasaya arz edilen ve halen piyasada bulunan tıbbi cihazların da piyasada kalmasına izin verecektir (yani, 'satış' tarihi yoktur).
Yönetmeliğin uygulanmasında kaydedilen önemli ilerlemeye rağmen, yeni düzenleyici çerçeveye başarılı bir geçiş sağlamak için
onaylanmış kuruluşların genel kapasitesi sınırlı kalmaktadır. Ayrıca birçok üretici, mevcut geçiş döneminin sonuna kadar Tıbbi
Cihazlar Yönetmeliği'nin sağlam gerekliliklerini karşılamaya yeterince hazır değildir. Bu durum, tıbbi cihazların AB pazarında
bulunabilirliğini tehdit etmektedir. Herhangi bir yasal işlem olmaksızın, piyasadaki çeşitli tıbbi cihazların tedariğinde
sağlık sistemini etkileyebilecek önemli aksaklıkların oluşması riski vardır.
Bu teklif, Tıbbi Cihazlar Yönetmeliği'nde öngörülen mevcut güvenlik ve performans gerekliliklerinin hiçbirini değiştirmez. Yalnızca,
imalatçılara önceden geçerli olan kurallardan Yönetmeliğin yeni gerekliliklerine geçişleri için daha fazla zaman tanımak amacıyla
geçiş hükümlerini değiştirir. Geçiş dönemleri için önerilen uzatma süresinin uzunluğu, cihazın türüne bağlıdır: kalp pili ve kalça
implantları gibi daha yüksek riskli cihazlar (Aralık 2027'ye kadar), şırıngalar veya yeniden kullanılabilir cerrahi aletler gibi
orta ve düşük riskli olanlara (Aralık 2028'e kadar) göre daha kısa bir geçiş döneminden faydalanacaktır.
Teklifin temel unsurları:
26 Mayıs 2021 tarihinden önce düzenlenmiş bir sertifika veya uygunluk beyanı kapsamındaki tıbbi cihazlar için yeni kurallara geçiş
süresi, daha yüksek riskli cihazlar için 26 Mayıs 2024'ten 31 Aralık 2027'ye, orta ve düşük riskli cihazlar için 31 Aralık 2028'e
kadar uzatılmıştır. . Uzatma belirli şartlara tabi olacak, böylece yalnızca güvenli olan ve üreticileri Tıbbi Cihazlar Yönetmeliği'nde
öngörülen kurallara geçiş için adım atmış olan cihazlar ek süreden yararlanabilecektir.
Teklif, sınıf III implante edilebilir ısmarlama cihazlar için de 26 Mayıs 2026'ya kadar bir geçiş dönemi getiriyor ve üreticilerine bir
onaylanmış kuruluş tarafından sertifika almaları için daha fazla zaman tanıyor. Ayrıca bu durumda geçiş süresi, imalatçının bu tip
cihazların uygunluk değerlendirmesi için 26 Mayıs 2024 tarihinden önce başvurusuna tabidir.
Teklif, bu değişikliklerle getirilen geçiş dönemlerini yansıtmak amacıyla, Tıbbi Cihazlar Yönetmeliği'nin yürürlüğe girdiği 26 Mayıs
2021 tarihine kadar düzenlenen sertifikaların geçerlilik sürelerini uzatıyor.
Komisyon ayrıca, Tıbbi Cihazlar Yönetmeliğinde ve Vücut Dışında Kullanılan Tıbbi Tanı Cihazları Yönetmeliğinde hâlihazırda belirlenmiş
olan 'satış' tarihinin kaldırılmasını teklif etmektedir. 'Satış' tarihi, hâlihazırda piyasaya arz edilmiş ve satın alınmaya hazır
durumda bulunan cihazların geri çekilmesi gereken bitiş tarihidir. Bu "satış" tarihinin kaldırılması, hâlihazırda piyasada bulunan
güvenli ve temel tıbbi cihazların sağlık hizmetleri sistemleri ve ihtiyacı olan hastalar tarafından kullanılabilir durumda kalmasını sağlayacaktır.
Teklifin şimdi hızlandırılmış bir ortak karar prosedürü yoluyla Avrupa Parlamentosu ve Konsey tarafından kabul edilmesi gerekiyor. Avrupa
Parlamentosu'nun 13-16 Şubat 2023 tarihlerinde yapılacak genel kurul oturumunda ilk okumada, Komisyon teklifinin metnini, acil durum
prosedürü yoluyla ilk okumadaki şekliyle, değişiklik yapılmadan oylaması bekleniyor.
Lütfen MDR Madde 10’ a bakın.
“Yetkili Temsilci”, Birlik dışında yerleşik bir imalatçıdan MDR kapsamındaki yükümlülüklerine ilişkin olarak belirtilen görevlerle ilgili olarak imalatçı adına hareket etmek üzere yazılı bir vekâlet almış ve kabul etmiş, Birlik içinde yerleşik herhangi bir gerçek veya tüzel kişi anlamına gelir. AR'nin sorumlulukları MDR'nin 11. maddesinde açıklanmıştır.
Bir yetkili temsilci değişimi için ayrıntılı düzenlemeler, imalatçı, uygulanabilir olduğu yerde ayrılan yetkili temsilci ve yeni başlayan yetkili temsilci arasındaki bir sözleşmede açıkça tanımlanır. Lütfen bu anlaşmanın gereklilikleri için MDR madde 12'ye bakın.
Yönetmelik aynı zamanda ithalatçıların görev ve sorumluluklarını da tanımlamaktadır. İthalatçı, üçüncü bir ülkeden bir cihazı AB pazarına arz eden, AB'de yerleşik herhangi bir gerçek veya tüzel kişi olarak tanımlanır. MDR Madde 13, ithalatçıların genel yükümlülüklerinin çoğunu açıklamaktadır.
Distribütör, tedarik zincirinde yer alan, imalatçı veya ithalatçı dışında, bir cihazı hizmete sunma anına kadar piyasada bulunduran herhangi bir gerçek veya tüzel kişi olarak tanımlanmaktadır. Yönetmelik, dağıttıkları cihazların MDR Madde 14'te açıklanan yükümlülüklere uygun olduğundan temsili örnekleme yoluyla emin olması gereken dağıtıcıların rollerini ve sorumluluklarını tanımlar.
MDR Madde 15 uyarınca, İmalatçılar; tıbbi cihazlar alanında gerekli uzmanlığa sahip, mevzuata uyumdan sorumlu en az bir kişiyi
kuruluşlarının bünyesinde bulundurur. Gerekli uzmanlık, aşağıdaki yeterliliklerden herhangi biri yoluyla kanıtlanır:
(a) hukuk, tıp, eczacılık, mühendislik veya ilgili başka bir bilimsel disiplinde, bir üniversite derecesi veya ilgili üye devlet tarafından
bunlara denkliği kabul edilen bir eğitimin tamamlanması üzerine verilen bir diploma, sertifika veya diğer resmi yeterlilik kanıtı ve
tıbbi cihazlarla ilgili, mevzuat işlerinde ya da kalite yönetim sistemlerinde asgari bir yıllık mesleki deneyim;
(b) tıbbi cihazlarla ilgili mevzuat işlerinde veya kalite yönetim sistemlerinde dört yıllık mesleki deneyim.
Mesleki yeterliliklerle ilgili ulusal hükümlere halel getirmeksizin ısmarlama üretilen cihazların imalatçıları, birinci alt paragrafta
atıfta bulunulan gerekli uzmanlıklarını, ilgili imalat alanında asgari iki yıllık mesleki deneyim ile kanıtlayabilir.
Mevzuata uyumdan sorumlu kişi asgari olarak şunları sağlamaktan sorumlu olur:
(a) bir cihaz piyasaya serbest bırakılmadan önce cihazların uygunluğunun bu cihazların imal edildikleri kalite yönetim sistemi uyarınca kontrol edilmesini,
(b) teknik dokümantasyonun ve AB uygunluk beyanının düzenlenmesini ve güncel tutulmasını,
(c) 10 (10) maddesi uyarınca piyasaya arz sonrası gözetim yükümlülüklerine uyulmasını,
(d) 87 ila 91. maddelerde atıfta bulunulan raporlama yükümlülüklerinin yerine getirilmesini,
(e) araştırma amaçlı cihazlar olması durumunda, XV. Ekin II. Bölümünün 4.1. Kesiminde atıfta bulunulan beyanın düzenlenmesi.
Madde 15’ in açıklığa kavuşturulması ile ilgili ayrıntılı bilgi için bkz.
MDCG 2019-7 ‘Mevzuata uyum sorumlusuna’ (PRRC) ilişkin Tıbbi Cihaz Tüzüğü (MDR) ve İn Vitro Tanı Tıbbi Cihazları Tüzüğü (IVDR) Madde 15 Hakkında Rehber
Hayır, 2003/361/AT sayılı komisyon tavsiyesi çerçevesindeki mikro ve küçük işletmelerin, mevzuata uyumdan sorumlu kişiyi
kendi kuruluşları bünyesinde bulundurmaları gerekmez; fakat söz konusu işletmeler kalıcı ve sürekli olarak böyle
bir kişiden hizmet alırlar.
İncelemek için
Tek kullanımlık cihazların yeniden işlenmesi ve daha fazla kullanımı, yalnızca ulusal yasaların izin verdiği durumlarda ve yalnızca
MDR Madde 17 uyarınca gerçekleştirilebilir.
Tek kullanımlık cihazların yeniden işlenmesine ilişkin ulusal kurallar hakkında daha fazla bilgi edinmek için aşağıdaki linki ziyaret edin.
Bir sağlık kurumu bünyesinde yeniden işlenen ve kullanılan tek kullanımlık cihazlar hususunda, aşağıdakilerin sağlanması şartıyla,
üye devletler, imalatçının bu Tüzük’te belirtilen yükümlülükleriyle ilgili kuralların bazılarını uygulamamaya karar verebilir:
yeniden işlenen cihazın güvenlilik ve performansının orijinal cihazınkine denk olması
yeniden işleme, risk yönetimi, tüm süreç için prosedürlerin doğrulanması, ürün serbest bırakılması ve
performans testi, kalite yönetim sistemi, olayların raporlanması ve izlenebilirlik ile ilgili gereklilikleri
detaylandıran Komisyon Uygulama Yönetmeliği (AB) 2020/1207'ye uygun olarak gerçekleştirilmesi.
Tek kullanımlık cihazların yeniden işlenmesine ilişkin ulusal kurallar
Komisyon Uygulama Yönetmeliği (AB) 2020/1207 için Tıklayınız
İmplant kartı, cihazı açıkça tanımlamalı ve ilgili ek bilgileri sağlamalıdır:
Cihaz adı.
Seri numarası veya lot veya parti numarası.
İnsan ve makine tarafından okunabilen formatta benzersiz cihaz tanımlaması (UDI).
Üreticinin adı, adresi ve web sitesi.
Üreticinin web sitesi.
Cihaz tipi.
Ek olarak, hasta ve sağlık hizmeti sağlayıcısı implant kartında tanımlanmalıdır:
Hastanın adı veya hasta kimliği.
İmplantasyonu gerçekleştiren sağlık kuruluşunun adı ve adresi.
İmplantasyon tarihi.
Sembollerin bu verileri iletmesine izin verilir. Bazı yeni semboller tanımlanmıştır:
Hasta adı veya hasta kimliği.
implantasyon tarihi.
İmplantı uygulayan sağlık kuruluşunun/sağlayıcının adı ve adresi.
Hastalar için bilgi sitesi.
Cihaz adı.
Bir implant kartının neleri içermesi gerektiği MDCG 2019-18 kılavuz belgesinde açıklanmaktadır.
Kılavuza ulaşmak için tıklayınız
MDR'nin 19. Maddesi uyarınca, AB uygunluk beyanı, kapsanan cihazla ilgili olarak bu Tüzük’te belirtilen gerekliliklerin yerine
getirildiğini belirtir. İmalatçı, AB uygunluk beyanını sürekli olarak günceller. AB uygunluk beyanı, asgari olarak IV. Ekte
belirtilen bilgileri içerir ve cihazın bulundurulduğu üye devlet(ler) tarafından talep edilen bir resmi birlik diline veya dillerine çevrilir.
AB uygunluk beyanı, Ek IV'e göre aşağıdaki bilgilerin tümünü içerecektir:
1. İmalatçının ve mevcutsa, yetkili temsilcisinin adı, kayıtlı ticari unvanı veya kayıtlı ticari markası ve hâlihazırda düzenlenmişse,
31. maddede atıfta bulunulduğu şekilde MKN’si ve kendilerine ulaşılabilecek ve konumları belirlenebilecek kayıtlı iş yerlerinin adresi;
2. AB uygunluk beyanının, imalatçının tamamen kendi sorumluluğu altında düzenlendiğine ilişkin bir beyan;
3. VI. Ekin C Kısımında atıfta bulunulduğu şekilde Temel UDI-DI;
4. AB uygunluk beyanının kapsadığı cihazın tanımlanmasına ve izlenebilirliğine olanak sağlayan, ürün adı ve ticari adı, ürün kodu, katalog
numarası veya uygulanabilir olduğu hallerde fotoğraf gibi diğer kesin referanslar ile birlikte kullanım amacı. Ürün adı veya ticari
adı hariç olmak üzere, tanımlamaya ve izlenebilirliğe olanak sağlayan bilgiler 3. bentte atıfta bulunulan Temel UDI-DI ile sağlanabilir;
5. VIII. Ekte belirtilen kurallar uyarınca cihazın risk sınıfı;
6. Mevcut beyan kapsamında olan cihazın bu Tüzük’e ve mevcutsa, bir AB uygunluk beyanının düzenlenmesini şart koşan ilgili diğer Birlik
mevzuatına uygun olduğuna dair bir beyan;
7. Kullanılan ve uygunluğun beyan edildiği ortak spesifikasyonlara (OS) ilişkin atıflar;
8. Uygulanabildiği hallerde, onaylanmış kuruluşun adı ve kimlik numarası, yürütülen uygunluk değerlendirme işleminin bir tanımı ve
düzenlenen sertifika veya sertifikaların tanımlanması;
9. Uygulanabildiği hallerde ilave bilgiler;
10. Beyanın düzenlenme yeri ve tarihi, imzalayan kişinin adı ve görevi ile birlikte bu kişinin kimin adına imzaladığının bilgisi, imza.